Mutation refers to a permanent change in the nucleotide sequence of an organism’s DNA. These changes can occur either naturally during DNA replication or as a result of environmental factors like ultraviolet (UV) radiation, ionizing radiation, and exposure to chemical agents. Mutations can disrupt the normal structure and function of DNA, affecting gene expression, protein synthesis, and ultimately, cellular function.
To combat these harmful alterations, cells are equipped with sophisticated and highly regulated systems capable of detecting and repairing DNA damage. Without these repair systems, persistent DNA damage can lead to severe biological consequences. These include the development of mutations, the onset of cancer, programmed cell death (apoptosis), premature aging, and genetic abnormalities.
Both endogenous and exogenous agents contribute to DNA damage. Internally, reactive oxygen species produced during metabolism and errors during DNA replication introduce lesions. Externally, factors like UV radiation, X-rays, and mutagenic chemicals pose constant threats to DNA integrity. Given the frequency and diversity of DNA damage, cellular DNA repair mechanisms play a crucial role in preserving genetic stability and overall health.
Summary of DNA Repair
- DNA repair preserves genetic stability by fixing damage from internal and external sources.
- Key pathways include Direct Reversal, BER, NER, MMR, DSB Repair, and TLS for different damage types.
- Faulty repair systems cause genomic instability and are linked to cancer and genetic diseases.
Table of Contents
Types of DNA Damage
Single Base Modifications
Single base modifications involve chemical alterations to individual bases in the DNA molecule. One common example is deamination, where cytosine is converted into uracil, resulting in incorrect base-pairing during replication. Oxidation is another type, where reactive oxygen species can modify guanine to form 8-oxoguanine, which mispairs with adenine instead of cytosine. Additionally, alkylation involves the addition of alkyl groups, such as methyl or ethyl groups, to DNA bases like guanine, potentially leading to abnormal base-pairing and mutations.
Structural Distortions
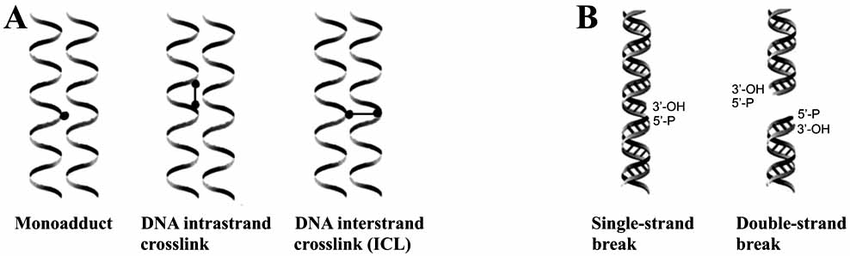
Structural distortions occur when the physical shape of the DNA double helix is disrupted, hindering essential processes like replication and transcription. Exposure to ultraviolet (UV) light can cause adjacent pyrimidine bases, such as thymine, to form abnormal covalent bonds called pyrimidine dimers, which bend the DNA strand. Carcinogenic chemicals like benzo[a]pyrene, found in cigarette smoke, can attach to DNA bases, forming bulky adducts that distort the helix. These structural changes interfere with the normal function of DNA polymerases and activate DNA repair mechanisms.
Strand Breaks
Strand breaks involve the breaking of the DNA’s sugar-phosphate backbone, either on one strand or both. Single-strand breaks (SSBs) affect only one of the two strands and are usually repaired efficiently by cellular repair systems. In contrast, double-strand breaks (DSBs) occur when both strands are severed, often due to ionizing radiation, oxidative stress, or replication errors, posing a serious threat to genome stability. If not properly repaired, DSBs can lead to chromosomal rearrangements or cell death.
Mismatched Bases
Mismatched bases arise when incorrect nucleotide pairs form during DNA replication, disrupting the normal base-pairing rules. For example, adenine might mistakenly pair with cytosine instead of its correct partner, thymine. If these errors are not corrected before the next round of replication, they can become permanent mutations in the DNA sequence. The cell relies on a specialized system called mismatch repair (MMR) to detect and fix these mismatched bases, maintaining the accuracy of genetic information.
The Importance of DNA Repair
The continuous onslaught of DNA damage requires robust systems to detect and repair these lesions efficiently. Without such systems, mutations would accumulate, threatening the cell’s viability and the organism’s health. DNA repair mechanisms are essential for preserving genetic information, preventing mutations from becoming fixed in the genome, and ensuring accurate transmission of genetic material to daughter cells during cell division.
These repair systems also enable the cell to differentiate between newly synthesized DNA and the original template strand during replication, ensuring that replication errors are corrected before becoming permanent mutations. Additionally, DNA repair prevents harmful chromosomal rearrangements such as translocations, deletions, and duplications, which are often implicated in cancer and genetic diseases.
Some DNA repair pathways restore the original sequence perfectly, making them error-free, while others are inherently error-prone, potentially introducing mutations during the repair process. Both types, however, are crucial for cell survival, particularly under conditions of extensive DNA damage.
Overview of DNA Repair Mechanisms
Cells employ multiple DNA repair mechanisms, each tailored to a specific type of damage and active at particular stages of the cell cycle. These systems differ in their enzymatic machinery, substrates, and fidelity.
Direct repair mechanisms directly reverse the chemical modifications affecting nucleotide bases. Excision repair systems, including base excision repair (BER) and nucleotide excision repair (NER), involve the removal of damaged regions followed by DNA resynthesis. Mismatch repair (MMR) detects and corrects errors introduced during DNA replication.
For double-strand breaks, cells rely on homologous recombination (HR) or non-homologous end joining (NHEJ), depending on the cell cycle phase and the availability of a homologous template. When damage occurs during DNA replication and cannot be repaired immediately, translesion synthesis (TLS) allows specialized polymerases to bypass lesions, although at the cost of reduced fidelity.
Each of these repair systems plays a distinct but complementary role in maintaining genomic integrity and preventing disease.
Direct Reversal of DNA Damage
Direct reversal represents the simplest and most energy-efficient DNA repair strategy. Instead of removing and replacing damaged bases, this mechanism restores the original chemical structure. This mechanism directly reverses the damage without replacing the DNA strand.
Photoreactivation
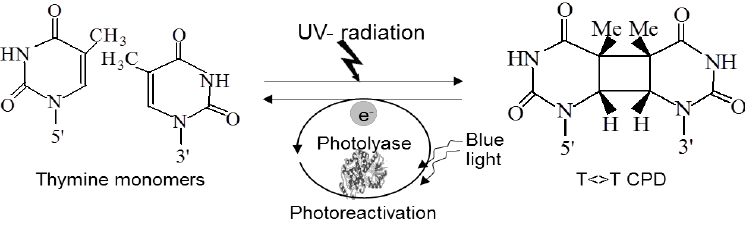
Photoreactivation is a direct DNA repair mechanism that corrects pyrimidine dimers, which form when adjacent pyrimidine bases bond abnormally due to ultraviolet (UV) light exposure. This repair is performed by an enzyme called photolyase, which recognizes and binds to the dimer. Activated by visible light, photolyase uses light energy to break the covalent bonds of the dimer, restoring normal base pairing. This repair system is present in many organisms like bacteria, plants, and fungi, but is absent in placental mammals, including humans.
Methyltransferases
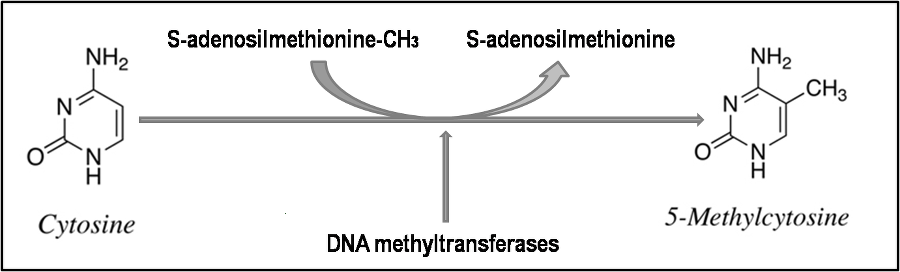
Methyltransferases are direct repair enzymes that remove unwanted methyl groups attached to DNA bases. O6-methylguanine-DNA methyltransferase (MGMT) is a key example, repairing O6-methylguanine, a mutagenic lesion on guanine bases. MGMT transfers the methyl group to a cysteine residue within its own structure, which inactivates the enzyme in a one-time, suicide reaction, requiring constant synthesis of new MGMT molecules to maintain DNA protection.
Excision Repair Pathways
Excision repair pathways are essential DNA repair mechanisms that remove damaged or abnormal bases and replace them with the correct nucleotides. These pathways handle a wide range of DNA lesions, from small base changes to bulky, helix-distorting damage. There are two major types: Base Excision Repair (BER) and Nucleotide Excision Repair (NER).
Base Excision Repair (BER)
Base Excision Repair (BER) corrects small, non-helix-distorting base lesions, such as those caused by deamination, oxidation, or alkylation. It maintains genomic stability by recognizing and repairing single altered bases before they cause mutations. BER is especially important for fixing spontaneous DNA damage caused by normal cellular metabolism.
Steps in Base Excision Repair (BER)
- Damage Recognition: A specific enzyme called DNA glycosylase detects and removes the damaged base from the DNA molecule by cleaving the bond between the base and the sugar, leaving the sugar-phosphate backbone intact.
- AP Site Formation: The removal of the base creates an abasic site (also called an AP site), where a base is missing but the sugar-phosphate backbone remains.
- Backbone Cleavage: An enzyme known as AP endonuclease cleaves the DNA backbone at the AP site, generating a nick in the strand.
- Nucleotide Insertion: The resulting gap is filled by DNA polymerase β, which inserts the correct nucleotide using the complementary DNA strand as a template.
- Ligation: Finally, the nick in the sugar-phosphate backbone is sealed by DNA ligase III, in cooperation with the XRCC1 protein, restoring the DNA to its original, undamaged state.
Key enzymes involved in BER:
- Uracil-DNA glycosylase (UNG)
- AP endonuclease
- DNA polymerase β
- DNA ligase III/XRCC1 complex
Nucleotide Excision Repair (NER)
Nucleotide Excision Repair (NER) removes bulky, helix-distorting DNA lesions, such as UV-induced thymine dimers and large chemical adducts. It is versatile because it recognizes a wide range of DNA damage that affects the shape of the double helix. NER operates through two sub-pathways: Global Genomic NER (GG-NER), which scans the entire genome, and Transcription-Coupled NER (TC-NER), which focuses on active, transcribed genes.
Steps in Nucleotide Excision Repair (NER)
- Damage Recognition: Specialized proteins recognize the distortion in the DNA helix caused by bulky lesions.
- DNA Unwinding: A complex called TFIIH uses its helicase activity to unwind the DNA around the damaged site, exposing the lesion.
- Dual Incision: Endonucleases make two cuts in the damaged strand: one on either side of the lesion, typically removing a segment of about 24–32 nucleotides.
- Excision of Damaged Strand: The damaged DNA fragment containing the lesion is then removed from the helix.
- Gap Filling: DNA polymerase δ or ε synthesizes new DNA using the undamaged strand as a template, filling in the gap.
- Ligation: Finally, DNA ligase I (or DNA ligase III in some cases) seals the remaining nick, completing the repair.
Disorders associated with NER deficiencies
- Xeroderma pigmentosum (XP): Caused by defects in GG-NER, leading to extreme UV sensitivity and skin cancer risk.
- Cockayne syndrome: Resulting from defects in TC-NER, characterized by growth failure, neurological degeneration, and sensitivity to sunlight.
Mismatch Repair (MMR)
Mismatch repair (MMR) corrects errors introduced during DNA replication, such as base mismatches and small insertion-deletion loops. This system is crucial for maintaining replication fidelity.
The repair process begins with damage recognition. The MSH2-MSH6 heterodimer (MutSα) detects base-base mismatches, while the MSH2-MSH3 complex (MutSβ) recognizes larger insertion-deletion loops. Once the mismatch is identified, the MLH1-PMS2 (MutLα) complex is recruited to the site.
The error-containing strand is then excised by exonucleases, and DNA polymerase δ synthesizes the replacement strand using the original template. The final step involves sealing the strand with DNA ligase, completing the repair.
In humans, defects in MMR genes lead to Lynch syndrome, also known as hereditary non-polyposis colorectal cancer (HNPCC), which significantly increases the risk of colorectal, endometrial, and other cancers.
Double-Strand Break Repair (DSBR)
Double-strand breaks (DSBs) represent one of the most lethal forms of DNA damage because they involve the simultaneous breakage of both strands of the DNA double helix. If left unrepaired, DSBs can cause chromosomal rearrangements, loss of genetic material, or cell death. To handle this critical damage, cells employ two primary repair mechanisms: Homologous Recombination (HR) and Non-Homologous End Joining (NHEJ).
Homologous Recombination (HR)
is an error-free repair pathway active predominantly in the late S and G2 phases of the cell cycle, when a sister chromatid is available to serve as an undamaged template. The repair process starts when sensor proteins detect the DSB and recruit the MRN complex, composed of MRE11, RAD50, and NBS1. This complex processes the DNA ends, creating 3’ single-stranded overhangs through resection. The RAD51 protein then facilitates the invasion of these overhangs into the homologous sequence on the sister chromatid, forming a displacement loop (D-loop). DNA polymerase extends the invading strand using the undamaged template. Finally, the structures are resolved, restoring the integrity of the DNA without loss of genetic information.
Non-Homologous End Joining (NHEJ)
in contrast, is an error-prone mechanism that directly ligates the broken DNA ends without the need for a homologous template. This pathway operates throughout the cell cycle but is especially important in G1 when no sister chromatid is present. The process begins with the binding of Ku70/Ku80 heterodimers to the DNA ends, protecting them from degradation. DNA-PKcs (DNA-dependent protein kinase catalytic subunit) is then recruited to align the ends, which are processed if necessary by nucleases like Artemis to create ligatable ends. Finally, DNA ligase IV, in association with XRCC4 and XLF, seals the break. Though efficient, NHEJ can lead to small insertions or deletions, potentially causing mutations.
Both HR and NHEJ are essential for maintaining genomic stability. Defects in these pathways are implicated in immunodeficiency syndromes and predisposition to cancer, highlighting the importance of tightly regulated double-strand break repair
Translesion DNA Synthesis (TLS)
When cells encounter lesions during DNA replication that cannot be immediately repaired, they risk stalling the replication machinery, potentially leading to replication fork collapse and double-strand breaks. To prevent such outcomes, cells employ a damage-tolerance mechanism known as Translesion DNA Synthesis (TLS).
TLS involves specialized DNA polymerases capable of synthesizing DNA across damaged templates. Unlike replicative polymerases, these TLS polymerases have flexible active sites that accommodate distorted or chemically modified bases. However, this flexibility comes at the cost of reduced fidelity and an increased risk of incorporating incorrect nucleotides.
When a replicative polymerase encounters a lesion, the replication fork stalls, triggering the recruitment of TLS polymerases such as Pol η, Pol ι, Pol κ, and REV1. Pol η is particularly notable for its ability to accurately bypass UV-induced thymine dimers, inserting the correct adenines opposite the dimer. Other polymerases, like Pol ι and Pol κ, are more error-prone but are essential for bypassing a variety of other lesions.
After bypassing the lesion, TLS polymerases are typically replaced by the high-fidelity replicative polymerases, and replication proceeds. This switching mechanism ensures that the damage is bypassed while limiting the extent of error-prone synthesis. While TLS helps cells survive DNA damage, its inherent mutagenicity contributes to the formation of mutations, underscoring a trade-off between cell survival and genome fidelity.
DNA Damage Checkpoints
Besides repair systems that directly correct DNA damage, cells employ surveillance mechanisms known as DNA damage checkpoints. These checkpoints halt cell cycle progression in response to DNA damage, providing time for repair before the cell continues through the cycle. Failure to properly activate these checkpoints can result in the propagation of mutations, genomic instability, and tumorigenesis.
Checkpoints operate at several stages of the cell cycle: G1/S, intra-S, and G2/M. The G1/S checkpoint ensures that damaged DNA is repaired before the cell commits to DNA replication. When damage is detected, proteins such as ATM (ataxia-telangiectasia mutated) and ATR (ATM and Rad3-related) are activated. These kinases phosphorylate downstream targets, including the tumor suppressor p53, which induces the expression of p21. The p21 protein inhibits cyclin-dependent kinases, halting the cell cycle.
The intra-S checkpoint slows replication in response to damage during the S phase. ATM and ATR also regulate this checkpoint by stabilizing replication forks and preventing the initiation of new replication origins.
At the G2/M checkpoint, cells verify the completion of DNA replication and the repair of any remaining damage before entering mitosis. If damage persists, checkpoint kinases CHK1 and CHK2 phosphorylate and inhibit CDC25 phosphatases, preventing activation of cyclin-dependent kinases necessary for mitotic entry.
In cases where damage is irreparable, checkpoints can trigger apoptosis, permanently removing damaged cells from the population. This coordination between repair systems and checkpoints is vital for preserving genomic integrity and preventing diseases like cancer.
Consequences of Defective DNA Repair
Defects in DNA repair pathways can have severe biological consequences. Inherited mutations in genes encoding repair proteins lead to syndromes characterized by cancer predisposition, premature aging, neurodegeneration, and immunodeficiency.
One example is Xeroderma pigmentosum (XP), caused by mutations in nucleotide excision repair genes. Patients with XP exhibit extreme sensitivity to UV light, a high incidence of skin cancers, and, in some cases, neurological abnormalities. Similarly, Lynch syndrome, resulting from defects in mismatch repair genes like MLH1, MSH2, or MSH6, predisposes individuals to colorectal and endometrial cancers.
Ataxia-telangiectasia (A-T) is another disorder arising from mutations in the ATM gene, which plays a key role in double-strand break repair and checkpoint control. A-T patients suffer from neurodegeneration, immunodeficiency, and increased cancer risk. Defects in homologous recombination genes, including BRCA1 and BRCA2, significantly elevate the risk of breast and ovarian cancers.
The cumulative failure of repair pathways contributes to genomic instability, a hallmark of cancer cells. Tumors often display mutations in multiple DNA repair genes, leading to a high mutation burden and chromosomal rearrangements that drive cancer progression.
Conclusion
The genome constantly faces damage from internal and external factors, risking mutations and diseases like cancer. To protect against this, cells use specialized DNA repair systems. Different pathways address specific damage types: direct reversal fixes base modifications, base and nucleotide excision repair handle small lesions and bulky distortions, mismatch repair corrects replication errors, and double-strand breaks are repaired by homologous recombination or non-homologous end joining.
Additional processes like translesion synthesis and checkpoints support genome stability. Defects in these systems can lead to disease, but also offer targets for therapies, such as PARP inhibitors for BRCA-related cancers. DNA repair is essential for preserving life and genetic fidelity.
Frequently Asked Questions (FAQ)
What are the methods of DNA repair?
Methods of DNA repair include direct repair, base excision repair, nucleotide excision repair, mismatch repair, double-strand break repair, and translesion synthesis.
How can mutations be repaired?
Mutations can be repaired by recognizing abnormal DNA structures or mismatched bases and restoring the correct sequence through these DNA repair pathways.
Can you fix a mutation.
While some mutations can be fixed by natural repair systems, others become permanent if not corrected before DNA replication.


