
Alpha-thalassemia is a genetic blood condition that impairs the production of hemoglobin, the protein in red blood cells that transports oxygen throughout the body. This disorder is caused by mutations or deletions in the HBA1 and HBA2 genes, which are required to produce alpha-globin chains, a critical component of hemoglobin. Understanding it involves thoroughly examining its causes, symptoms, kinds, diagnosis, and treatment.
Table of Contents
Causes of Alpha-Thalassemia
It is mostly caused by genetic mutations. Humans have four alpha-globin genes, with two on each chromosome 16. The severity of the condition is directly proportional to the number of these genes impacted.
Silent Carrier State: A mutation in a single gene. People are usually asymptomatic and may be unaware of their carrier status. This disorder does not create any health consequences and is often detected through genetic testing.
The Alpha-Thalassemia Trait: Results from mutations in two genes. This illness can produce moderate anemia, however, it generally goes untreated due to the lack of major symptoms. Individuals with this trait may have modest hemoglobin decreases, although they normally live normal lives without needing therapy.
Hemoglobin H Disease: Caused by three genetic mutations. It causes moderate to severe anemia, jaundice, splenomegaly (an enlarged spleen), bone abnormalities, tiredness, and growth retardation. This type has a major impact on quality of life and requires ongoing medical care and control.
Alpha-Thalassemia Major: Also known as hydrops fetalis, is a severe condition caused by abnormalities in all four alpha-globin genes. If not treated promptly, it usually leads to fetal mortality or serious health issues shortly after birth. Babies with this syndrome cannot make functional hemoglobin, resulting in severe anemia and organ failure.
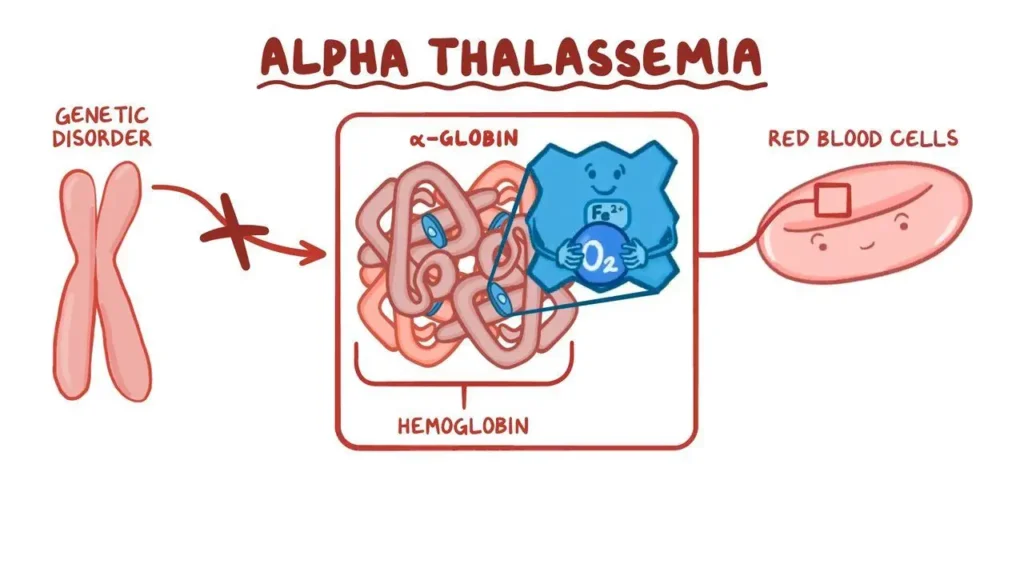
Types
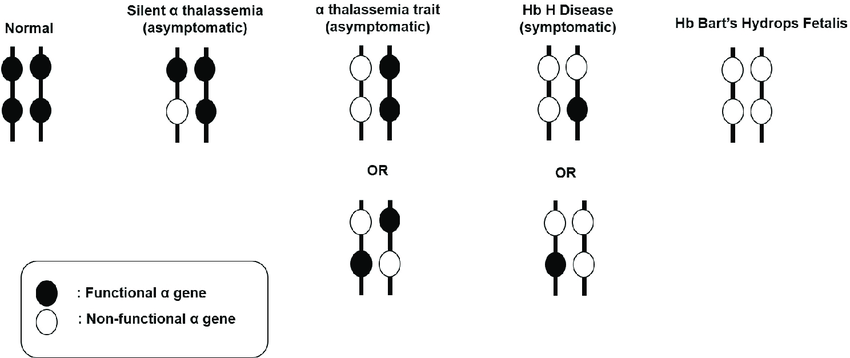
It is classified based on the number of alpha-globin genes affected by mutations or deletions.
1. Alpha-Thalassemia Silent Carrier
- One gene is missing or damaged.
- Symptoms: Usually no symptoms. Blood tests are often normal.
- Inheritance: Can pass the damaged gene to children.
2. Alpha-Thalassemia Trait
- Two genes are missing.
- Symptoms: Mild anemia may occur but often no symptoms.
- Inheritance: Can pass the damaged gene to children.
3. Hemoglobin H Disease
- Three genes are missing.
- Symptoms: Moderate to severe anemia.
- Treatment: May require frequent blood transfusions.
4. Alpha-Thalassemia Major (or Hydrops Fetalis)
- All four genes are missing.
- Symptoms: Severe anemia, often fatal before or shortly after birth.
- Treatment: Requires immediate medical attention.
Symptoms
The symptoms can vary greatly depending on the kind and severity of the illness.
Silent Carrier State: Individuals normally have no symptoms and live regular lives. This issue is frequently diagnosed through genetic testing or when a family history of thalassemia is examined.
Alpha-Thalassemia Trait: Symptoms are often modest and may include minor anemia. Individuals may experience slight weariness, but they are unlikely to have any serious health complications. Routine blood tests may reveal microcytic anemia, a condition in which red blood cells are smaller than normal.
Hemoglobin H Disease: Symptoms include moderate to severe anemia, jaundice (yellowing of the skin and eyes), splenomegaly, bone abnormalities (particularly in the face and jaw), and weariness. In some situations, children may experience growth retardation and delayed development. Patients may also have hemolysis, which occurs when red blood cells are destroyed quicker than they are generated.
Alpha-Thalassemia Major: A disorder that causes severe anemia, widespread swelling (hydrops fetalis), and heart failure in the fetus. Without intervention, it frequently results in stillbirth or early neonatal mortality. Babies born with this syndrome require early medical care to treat their severe anemia and related problems.
Diagnosis
It is diagnosed through a combination of blood tests and genetic testing:
A complete blood count (CBC): Can detect microcytic anemia (tiny red blood cells). This test is frequently the first sign that a person may have thalassemia, requiring additional study.
Hemoglobin electrophoresis: Detects aberrant hemoglobin types. This test distinguishes this disease from other kinds of anemia and verifies the existence of defective hemoglobin variations.
Genetic testing: Confirms the presence of mutations or deletions in alpha-globin genes. This conclusive test identifies the particular genetic alterations causing the illness and is critical for correct diagnosis and family planning.
Prenatal testing: Includes chorionic villus sampling (CVS) or amniocentesis for high-risk pregnancies. These tests can detect alpha-thalassemia in fetuses, allowing for timely intervention and planning.
Treatment
The treatment for alpha-thalassemia depends on its severity:
Silent Carrier and Alpha-Thalassemia Trait: These conditions usually do not require treatment. These people lived typical lives with no major health difficulties.
Hemoglobin H Disease: Treatment may include regular blood transfusions, folic acid supplements, and, in extreme cases, a splenectomy (splenectomy) or bone marrow transplant. Regular monitoring and management of anemia and accompanying consequences is critical for preserving quality of life.
Alpha-Thalassemia Major: Requires rigorous treatment, including regular blood transfusions, iron chelation therapy to prevent iron overload from transfusions, and maybe a bone marrow or stem cell transplant, as the only curable option. To address the severe complications of this illness, patients must receive lifelong medical care and monitoring.
To summarize, while alpha-thalassemia is a complex and varied illness, advances in medical care and genetic understanding have greatly improved the prognosis for many people. Ongoing research is investigating better treatment options and potential remedies for this genetic illness. Understanding the genetic foundation, detecting symptoms early, and implementing suitable treatment techniques are critical to properly treating this disease and allowing affected persons to live healthy, fulfilling lives.
Frequently Asked Questions (FAQ)
What is alpha-thalassemia?
Alpha-thalassemia is a genetic blood condition that impairs the production of hemoglobin, the protein in red blood cells that transports oxygen throughout the body.
How is alpha-thalassemia diagnosed?
Through blood tests (full blood count, hemoglobin electrophoresis), as well as genetic testing. Prenatal screening is also accessible for high-risk pregnancies.
How does alpha-thalassemia affect pregnancy?
Severe types, such as alpha-thalassemia major, can lead to serious pregnancy issues, including hydrops fetalis, which is deadly to the fetus.
Related Articles